


Les neurones humains dérivés d'iPSC révèlent une altération précoce de la croissance des neurites dans le syndrome de Wolfram, une maladie neurodégénérative tardive.
Sandra Pourtoy-Brasselet 3,6, Axel Sciauvaud 1,2,6, Maria-Gabriela Boza-Moran 1,2, Michel Cailleret 1,2, Margot Jarrige 1,2,3, Hélène Polvèche 3, Jérôme Polentes 3, Eric Chevet 4,5, Cécile Martinat 1,2, Marc Peschanski 1,2,3, Laetitia Aubry 1,2,*
1 INSERM UMR 861, I-STEM, AFM, Corbeil-Essonnes, 91100, France.
2 Université Paris-Saclay, Inserm, Univ Evry, Institut des Cellules souches pour le traitement et l'étude des maladies monogéniques, Corbeil-Essonnes, 91100, France.
3 CECS/AFM, I-STEM, Corbeil-Essonnes, 91100, France.
4 INSERM U1242, Université Rennes 1, Rennes, 35000, France.
5 Centre de Lutte Contre le Cancer Eugène Marquis, Rennes, 35000, France.
6 These two authors contributed equally to this work.
* Corresponding author: Laetitia Aubry, Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser. Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
RÉSUMÉ :
Des études récentes indiquent que les processus neurodégénératifs qui apparaissent pendant l'enfance et l'adolescence chez les personnes atteintes du syndrome de Wolfram (WS) s'ajoutent à l'altération précoce du développement cérébral, qui est cliniquement silencieuse. Les mécanismes pathologiques sous-jacents sont encore inconnus. Nous avons utilisé des cellules neurales dérivées de cellules souches pluripotentes induites provenant de personnes atteintes du syndrome de Wolfram afin de révéler leurs corrélats phénotypiques et moléculaires. Nous avons observé qu'une sous-population de neurones de Wolfram présentait une croissance neuritique aberrante associée à une expression altérée des gènes de guidage des axones. L'inhibition sélective du bras ATF6α de la réponse aux protéines mal conformées a permis de prévenir le phénotype altéré, bien que la réponse au stress aigu du réticulum endoplasmique - qui est activée dans les processus dégénératifs tardifs de Wolfram - n'ait pas été détectée. Parmi les médicaments actuellement testés chez les personnes atteintes de WS, l'acide valproïque est celui qui a permis de prévenir les phénotypes pathologiques. Ces résultats suggèrent que des défauts précoces dans le guidage des axones pourraient contribuer à la perte de neurones chez les personnes atteintes du syndrome de Wolfram.
INTRODUCTION
Le syndrome de Wolfram (WS, OMIM 222300) est une maladie monogénique multisystémique rare qui se caractérise par un ensemble de symptômes apparaissant pendant l'enfance et l'adolescence1. Des processus neurodégénératifs focaux se produisent alors, affectant les systèmes neurosensoriels et le tronc cérébral. Les résultats récents d'études d'imagerie par résonance magnétique ont toutefois révélé que les personnes atteintes du syndrome de Wolfram présentent en outre une réduction globale statistiquement significative du volume cérébral. L'origine de cette réduction du volume cérébral a été attribuée à des défauts de développement neurologique encore inconnus2-4 car elle est présente dès que les auteurs l'ont recherchée et reste stable, contrairement aux processus neurodégénératifs apparaissant plus tard et progressant avec le temps. Ce résultat pourrait être rapproché d'analyses plus anciennes de cerveaux de personnes atteintes du syndrome de Wolfram qui avaient mis en évidence une axonopathie généralisée5 ou une leucoencéphalopathie diffuse6 en l'absence de corrélats cliniques.
Les mécanismes moléculaires qui sous-tendent le syndrome de Wolfram, qui résulte de mutations du gène WFS1 (OMIM 606201) et par conséquent de la perte de la protéine wolframinee, ont été principalement attribués à un défaut dans la réponse au stress du réticulum endoplasmique (RE)7 ; 8. Ceci est soutenu par la localisation de la wolframinee à la membrane du RE9 et son rôle d'inhibiteur de la branche ATF6α de la réponse aux protéines non repliées10 (UPR). En conséquence, son absence dans les cellules β pancréatiques des individus atteints de WS a été associée à un contrôle défectueux de l'UPR conduisant au dysfonctionnement et à la mort cellulaire11, et au diabète. Des défauts dans la réponse au stress du RE ont également été proposés comme le principal mécanisme pathologique qui conduit aux processus neurodégénératifs progressifs qui affectent les neurones ganglionnaires de la rétine et, par extension, les neurones sensoriels de l'oreille et du tronc cérébral qui présentent également de fortes pertes au fil du temps12. Ces phénomènes étaient toutefois difficiles à concilier avec les caractéristiques spatiales et temporelles de la réduction globale du volume cérébral.
Dans la présente étude, nous avons cherché à étudier les mécanismes cellulaires et moléculaires associés à la pathologie neurodéveloppementale du syndrome de stress post-traumatique. Nous avons émis l'hypothèse que des neurones différenciés à partir de cellules souches pluripotentes induites (iPS) nous fourniraient un modèle expérimental directement pertinent afin de comparer les phénomènes neurodéveloppementaux précoces de donneurs sains et de donneurs Wolfram. Il a été démontré que les protocoles de différenciation des cellules souches pluripotentes basés sur la double inhibition des voies activin/nodal et BMP, les deux voies canoniques dépendantes de Smad, récapitulent bien la progression des cellules le long de la lignée neuronale13 ; 14. En particulier, les neurones corticaux glutamatergiques peuvent être spécifiquement différenciés15. Ceci nous a donc permis de comparer des populations neuronales similaires provenant de donneurs sains et de donneurs Wolfram à des stades précoces du développement neuronal et de révéler des anomalies pathologiques qui fournissent des indices pour comprendre les mécanismes qui conduisent aux défauts du volume cérébral identifiés chez les individus atteints de WS.
MATÉRIEL ET MÉTHODES
Reprogrammation des fibroblastes et culture de cellules souches pluripotentes
Les lignées iPSC de contrôle CT1 et CT3 ont été reprogrammées à I-Stem et déjà publiées15 ; 16 (CT1 correspondant à la lignée 1869 et CT3 à la lignée PB12). La lignée iPSC CT2 a été reprogrammée à partir de la cellule fibroblastique pulmonaire IMR-90 (ATCC® CCL-186). Les lignées iPSC WS1 et WS2 ont été aimablement données par la New York Stem Cell Foundation et ont déjà été publiées17. La lignée iPSC WS5 a été dérivée de fibroblastes fournis par le Dr Cécile Delettre et le regretté Pr Christian Hamel, isolés à partir d'un individu atteint de WS par biopsie après approbation du comité d'éthique local (NCT03853252, Institut des Neurosciences de Montpellier, France). Toutes les lignées cellulaires utilisées pour cette étude ainsi que les processus expérimentaux ont été réalisés conformément à la politique légale française. Par conséquent, le consentement éclairé a été obtenu de tous les participants ou de leurs représentants légaux.
Les fibroblastes ont été cultivés dans un milieu de culture pour fibroblastes constitué de DMEM high glucose (Invitrogen), GlutaMAX™ Supplement (Invitrogen), complété par 10% de sérum bovin fœtal (FBS, Sigma-Aldrich), 1% d'acides aminés non essentiels (Invitrogen) et 1 mM de pyruvate de sodium (Invitrogen). Les lignées d'iPSC CT2 et WS5 ont été obtenues et caractérisées comme décrit par Yu et al. 18 en utilisant les plasmides Addgene 20925, 20926 et 20927. Les iPSC ont été cultivées sur des MEF dans un milieu de culture de hESCs constitué de DMEM avec 20% de KSR, 1% d'acides aminés non essentiels, 1% de Glutamax, 0. 1% β-mercaptoéthanol et 0,1% pénicilline / streptomycine (tous de ThermoFisher Scientific) et complété avec 10 ng/mL FGF-2 (Peprotech) avec un passage manuel une fois par semaine et un changement de milieu quotidien pendant les étapes de caractérisation. Ils ont ensuite été adaptés à la culture sans nourrisseur et cultivés sur des plats recouverts de vitronectine (ThermoFisher Scientific) dans StemMACS™ iPS-Brew XF (Miltenyi Biotec). Les cultures ont été alimentées tous les deux jours et passées tous les 5 à 7 jours.
Différenciation des cellules souches pluripotentes
Pour la différenciation des corps embryoïdes (EBs), les colonies iPSC se développant sur des MEFs ont été détachées avec 1 mg/mL de collagénase (ThermoFisher Scientific) pendant 10 minutes à 37°C, remises en suspension dans un milieu hESCs sans FGF-2 et cultivées dans des plaques 6 puits à faible attachement pendant 7 jours à 37°C et 5% de CO2. Les EBs ont ensuite été placées sur des plaques recouvertes de gélatine à 0,1% (Sigma-Aldrich) et maintenues pendant 7 jours supplémentaires avant l'immunomarquage.
La différenciation des cellules souches neurales (NSCs) a été réalisée comme décrit précédemment15. À ce stade de la différenciation, les cellules peuvent être congelées dans une solution de sérum bovin fœtal 90%/10% diméthylsulfoxyde ≥99,5% (DMSO, 0,005%, VWR). Après décongélation, les cellules ont été ensemencées sur un support revêtu de poly-L-ornithine / laminine (PO/LAM), dans un milieu N2B27 constitué de DMEM/F12, de suppléments neurobasal, N2 et B27, de 0,1% de β-mercaptoéthanol et de 0,1% de pénicilline/streptomycine (tous de ThermoFisher Scientific) complété par 10 ng/mL de FGF-2 et EGF (Peprotech) et 10 µM de Y-27632 (Stemcell Technologies). Le milieu a été changé le jour suivant sans Y-27632 et ensuite changé tous les deux jours. Les NSCs ont été passées deux fois par semaine en utilisant Trypsin-EDTA 0,05 % (ThermoFisher Scientific) et ensemencées sur un support recouvert de PO/LAM à une densité de 50 000 cellules/cm² pour l'amplification.
La différenciation neuronale a été induite en plaçant les NSC à faible densité (50 000 cellules/cm²) dans des plaques de culture recouvertes de PO/LAM dans le milieu N2B27 sans FGF-2 et EGF. Au quatrième jour, 10 µM de DAPT (R&D Systems) ont été ajoutés au milieu pour arrêter la prolifération cellulaire. Le milieu a été changé tous les 4 jours et les neurones ont été obtenus après 18-21 jours de différenciation in vitro (DIV).
Traitements médicamenteux
Pour les expériences de traitements médicamenteux, les NSCs ont été différenciées en neurones comme décrit précédemment. Les neurones ont été traités deux fois à DIV 10 et DIV 14 avec les molécules et les concentrations suivantes : 19 : 10 µM de ceapin-A7 (Sigma-Aldrich) ; 2. 5 µM MKC-8866 (Medinnovata) ; 10 µM de salubrinal (Sigma-Aldrich) ; 100 nM de liraglutide (Bio-Techne) ; 10 µM de dantrolène (Sigma-Aldrich) ; 1 mM d'acide 4-phénylbutyrique (4PBA, Merck) ; 0,5 mM ou 1,5 mM d'acide valproïque (VPA, Sigma-Aldrich). La céapine-A7, le MKC-886, le salubrinal, le liraglutide et le dantrolène ont été remis en suspension dans du DMSO. Pour les analyses ultérieures, les cultures ont été arrêtées à DIV 18. La neuraminidase (NM) à 10 mU (Sigma-Aldrich) a été utilisée comme contrôle technique pour empêcher la fasciculation car cette enzyme élimine spécifiquement les résidus d'acide sialique et réduit par conséquent l'adhésion non spécifique entre les neurites20.
Kit d'expression génique du transcriptome humain Ion ampliseq
Pour chacun des 12 échantillons, 50 ng d'ARN total ont été transcrits de manière inverse à l'aide du kit Ion AmpliSeq Transcriptome Human Gene Expression suivant le protocole du fabricant (Thermofisher Scientific)21. Les bibliothèques d'ADNc ont été amplifiées et dotées d'un code-barres à l'aide du panneau central Ion AmpliSeq Transcriptome Human Gene Expression et de l'adaptateur de code-barres Ion Xpress (Thermofisher Scientific). Les amplicons ont été quantifiés à l'aide du kit ADN haute sensibilité d'Agilent avant que les échantillons ne soient regroupés en séries de huit. La PCR en émulsion et l'enrichissement ont été réalisée sur l'instrument Ion OT2 system en utilisant le kit Ion PI Hi-Q OT2 200 (Thermofisher Scientific). Les échantillons ont été chargés sur une puce Ion PI v3 et séquencés sur le système Ion Proton en utilisant la chimie du kit Ion PI Hi-Q sequencing 200 (longueur de lecture de 200 pb ; Thermofisher Scientific). Les lectures Ion Proton (fichiers FASTQ) ont été importées dans le pipeline RNA-seq du logiciel Partek Flow (v6 Partek Inc) en utilisant hg19 comme génome de référence. Le nombre de lectures par échantillon était compris entre 7,1 millions et 10 millions de lectures. Pour déterminer les gènes qui sont exprimés de manière différentielle entre les groupes, les lectures cartographiées ont été quantifiées à l'aide de l'algorithme E/M de Partek après normalisation par le nombre total d'échantillons (les comptes résultants représentent les niveaux d'expression des gènes sur les lectures/millions pour plus de 20 800 gènes différents présents dans la base de données de gènes humains (AmpliSeq Human Gene Expression panel). Les gènes exprimés de manière différentielle ont été identifiés à l'aide de l'algorithme Gene Specific Analysis (GSA) de Partek. Les listes de gènes ont été filtrées à pvalue ≤ 1% ;|Fold Change| ≥ 1,5 minReads100. L'utilisation de la pvalue au lieu de la pvalue ajustée est justifiée par la signification biologique et les validations expérimentales (Figure S4C)22. Les interprétations biologiques de la liste des gènes différentiellement exprimés ont été effectuées à l'aide de logiciels dédiés (Partek Genomics Suite (v6.6), et l'outil d'analyse d'enrichissement de la liste des gènes basé sur le Web EnrichR23 ; 24). Les analyses EnrichR ont été effectuées sur la base de données KEGG2016 et le processus biologique GO 2017b. Les données Ampliseq produites dans cette étude ont été déposées sur NCBI GEO sous le numéro d'accession GSE156911.
Imagerie time-lapse de la différenciation neuronale
Les NSC ont été ensemencées à 50 000 cellules/cm² dans des plaques de culture à 384 puits recouvertes de PO/LAM dans le milieu N2B27 pour induire une différenciation neuronale. Les plaques ont été incubées dans un système d'imagerie de cellules vivantes IncuCyte Zoom (Essen Bioscience) et des images en contraste de phase ont été prises automatiquement sous un grossissement microscopique de 10X toutes les 4 heures pendant les deux premières semaines de différenciation, puis toutes les 12 heures pendant les deux semaines suivantes. Les images ont été extraites et empilées à l'aide du logiciel IncuCyte Zoom pour créer les films.
Génération de la lignée d'iPSC WS5 « rescapée » (WS5R)
La lignée WS5R a été générée par knock-in médié par CRISPR-Cas9 pour introduire l'ADNc WFS1, sous un promoteur inductible par la doxycycline, au locus AAVS1. Comme backbone, nous avons utilisé le plasmide pAAVS1-PDi-CRISPRn (Addgene #73500). La cassette Cas9 a été remplacée par l'ADNc WFS1 du plasmide WFS1-pcDNA3 (Addgene#13011) en utilisant l'assemblage Gibson. Ce vecteur comprend des bras d'homologie dans les régions 5' et 3' flanquant immédiatement la séquence d'ADN du locus AAVS1 afin de diriger spécifiquement l'intégration du vecteur dans le locus AAVS1. Ce plasmide est principalement composé de rtTA sous le promoteur pCAG, de PuroR sous le contrôle du peptide T2A et du promoteur inductible TR3G qui permet l'expression de WFS1 avec un traitement à la doxycycline. Pour la génération de la lignée cellulaire de secours WS5, les cellules iPS WS5 ont d'abord été transfectées avec le plasmide linéarisé en utilisant Lipofectamine® 3000 (ThermoFisher Scientific). Les cellules ont été transfectées une seconde fois avec la RNP de nucléase/gRNA Cas9 préformée in vitro (Integrated DNA Technologies, IDT) en utilisant la Lipofectamine™ CRISPRMAX™ (ThermoFisher Scientific). La double transfection a été réalisée en suivant les protocoles du fabricant.
La formation du complexe RNP a été réalisée en utilisant l'ARN guide T2 (IDT) qui cible le locus AAVS1 : (GGGGCCACTAGGGACAGGAT) qui a été complexé avec la protéine spCas9 (ratio 1.2/1) pendant 10 minutes à température ambiante avant la transfection. Les clones ont été sélectionnés après une semaine de culture avec le milieu StemMACS™ iPS-Brew XF (Miltenyi Biotec) et 200 ng/mL de puromycine (ThermoFisher Scientific). Les iPSCs WS5R ont été différenciées en NSCs et en neurones. Afin d'induire la production de wolframine, les NSCs ont été traitées avec 50 ng/mL de doxycycline (Sigma-Aldrich) à chaque changement de milieu pendant 10 jours. Le traitement à 50 ng/mL de doxycycline a ensuite été maintenu tout au long de la différenciation en neurones.
Immunocytochimie
Les cellules ont été fixées avec du paraformaldéhyde à 4% (Electron Microscopy Sciences) pendant 15 minutes à température ambiante. Les cellules ont ensuite été placées dans un tampon de blocage (solution saline tamponnée au phosphate, 2 % d'albumine de sérum bovin et 0,1 % de Triton X-100). Les anticorps primaires ont été dilués dans le tampon bloquant et appliqués pendant la nuit à 4 °C. Les anticorps secondaires conjugués aux fluorophores Alexa (ThermoFisher Scientific) ont été dilués à 1:1000 dans un tampon de blocage et appliqués pendant 1 heure à température ambiante dans l'obscurité. Du Hoechst 33258 (Sigma-Aldrich) dilué au 1:3000 a été appliqué en même temps pour visualiser les corps cellulaires. Les cellules ont été visualisées sur un microscope inversé à fluorescence Zeiss et l'acquisition des images a été réalisée à l'aide du logiciel Zen Blue (Zeiss). Lorsqu'une résolution plus élevée était nécessaire, l'imagerie confocale a été réalisée à l'aide d'un microscope confocal Zeiss LSM880-Airyscan piloté par le logiciel Zeiss Zen black. Les anticorps utilisés sont décrits dans le tableau S2.
Trois différenciations biologiquement indépendantes pour chaque condition ont été étudiées et 5 images à grossissement 20X pour chaque différenciation ont été quantifiées. En raison du nombre élevé de fibres sur chaque image, la quantification de l'épaisseur des neurites a été abordée en utilisant une méthode d'échantillonnage aléatoire non biaisée dans ImageJ/FIJI. La méthode était basée sur l'utilisation d'une grille (surface par point : 8000 µm²) apposée automatiquement sur l'image par le logiciel. Un tiers aléatoire des carrés composant la grille a été quantifié pour chaque image. Sur chaque carré compté de la grille, l'épaisseur des neurites a été mesurée manuellement en utilisant l'outil de sélection d'imageJ pour délimiter le diamètre de chaque fibre. On a ensuite demandé à imageJ de mesurer la longueur de ligne correspondante (c'est-à-dire l'épaisseur de la fibre). Les résultats de chaque image ont ensuite été compilés sur un tableau global pour être classés en 3 intervalles d'épaisseur arbitraires (0 - 3 µm ; 3 - 6 µm ; > 6 µm). Les mêmes données ont été utilisées pour analyser l'épaisseur moyenne du réseau neuronal pour chaque condition.
Pour quantifier les proportions/pourcentages de cellules positives pour chaque marquage, la plateforme CellInsight CX7 High-Content Screening (HCS) et le logiciel HSC Studio (Thermofisher Scientific) ont été utilisés. Sur la base de la bioapplication CX7-Colocalization et de l'acquisition d'images à un grossissement de 10X, un algorithme maison avait permis la détection et la quantification individuelles des noyaux (Hoechst), du HuC/D (vert) et du CUX2 (rouge), et enfin le calcul de la proportion de chaque marquage par rapport au nombre total de cellules, comme lectures quantitatives.
Extraction de l'ARNm et PCR par transcription inverse (RT)
Les échantillons d'ARN total ont été isolés à l'aide du kit d'extraction RNeasy Mini ou Micro Plus et de l'instrument QIAcube (Qiagen) conformément au protocole du fabricant. La quantification et la qualité de l'ARN ont été évaluées à l'aide du spectrophotomètre NanoDrop ND-1000 (ThermoFisher Scientific) et du logiciel ND V33.7.1. Les échantillons d'ADNc ont été synthétisés à partir de 500 ng d'ARN avec la transcriptase inverse SuperScript III (ThermoFisher Scientific) en utilisant un mélange de 500 ng/µL d'amorces aléatoires (ThermoFisher Scientific) et 500 ng/µL d'Oligo(dT) (ThermoFisher Scientific).
PCR quantitative (qPCR)
Les tests de PCR quantitative ont été réalisés avec le mélange maître Luminaris HiGreen qPCR, low ROX (ThermoFisher Scientific) en utilisant le système QuantStudio 12K Flex Real-Time PCR (ThermoFisher Scientific), avec une étape de maintien (50°C pendant 2 minutes et 95°C pendant 10 minutes) suivie de 40 cycles de dénaturation (95°C pendant 15 secondes) et de recuit (60°C pendant 1 minute). La quantification de l'expression des gènes a été basée sur la méthode 2-ΔΔCt et normalisée à l'expression de 18S. Les amorces listées dans le tableau S3 ont été utilisées pour l'expression des gènes.
Traitement à la thapsigargine
Les NSC ont été ensemencées à 75 000 cellules/cm² sur des plaques de 6 puits recouvertes de PO/LAM dans du milieu N2B27 complété par 10 ng/mL d'EGF et de FGF-2 et incubées à 37°C, 5% CO2 pendant la nuit. Le jour suivant, les cellules ont été traitées avec l'inhibiteur non-compétitif de la sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 50 nM thapsigargin (TG, Sigma-Aldrich). Les cellules ont été recueillies à l'aide du tampon de lyse RLT+ (Qiagen) après 4 heures, 6 heures et 8 heures de traitement. Pour chaque point temporel, la condition témoin de DMSO a été collectée.
Analyse du gel XBP1
L'extraction de l'ARN et la RT ont été effectuées comme décrit ci-dessus. Pour l'analyse de l'épissage de XBP1, une amplification PCR a été réalisée avec 0,05 U/µL d'ADN polymérase Taq recombinante (ThermoFisher Scientific) et les amorces listées dans le tableau S3. L'amplification a été réalisée en utilisant une première étape à 95°C pendant 5 minutes, suivie de 30 cycles de dénaturation (60 secondes à 95°C), puis d'une étape d'annexion (30 secondes à 59°C) et enfin d'une étape d'élongation (30 secondes à 72°C), dans le Mastercycler Eppendorf®. Les produits PCR (XBP1 épissé (XBP1s) et XBP1 non épissé (XBP1u) respectivement à 193 pb et 223 pb) ont été quantifiés à l'aide du BioAnalyzer 2100 et du kit Agilent DNA 1000 (Agilent) selon le protocole du fabricant. L'expression de XBP1 épissé a été normalisée par rapport à XBP1 total (XBP1s + XBP1u).
Immunoblotting occidental
Les échantillons de protéines ont été isolés à partir de lysats cellulaires en utilisant le tampon RIPA (Sigma-Aldrich) complété par un cocktail d'inhibiteurs de protéase à 1 % (Sigma-Aldrich) et un inhibiteur de phosphatase à 10 % (Roche). La quantification des échantillons a été réalisée à l'aide du kit de dosage des protéines BCA de Pierce® (ThermoFisher Scientific) avec l'instrument CLARIOstar Plus (BMG Labtech). Les échantillons ont été préparés à une concentration de 30 μg de protéines et ont été placés 10 minutes à 70°C. Les protéines ont été chargées sur des gels Nu-PAGE® Bis-Tris à 4-12% (ThermoFisher Scientific) avec l'échelle de protéines pré-colorées Chameleon® Duo (Li-Cor) et ont migré pendant 10 minutes à 50 mA et 40 minutes à 100 mA. Les gels ont ensuite été transférés sur des membranes de nitrocellulose (ThermoFisher Scientific) à l'aide du système de transfert à sec iBlot 2 (ThermoFisher Scientific). Les buvards ont été bloqués pendant 1 heure à température ambiante dans le tampon de blocage Odyssey® (Li-Cor), puis incubés avec l'anticorps primaire dans le tampon de blocage avec 0,2 % de TWEEN 20 (Sigma-Aldrich) pendant la nuit à 4°C. Les blots ont ensuite été incubés avec des anticorps secondaires fluorescents (1:10 000, Li-Cor) dans le tampon bloquant avec 0,2% de TWEEN 20 (Sigma-Aldrich) dans l'obscurité à température ambiante pendant 1 heure. Les blots ont été révélés en utilisant le système d'imagerie Odyssey CLx (Li-Cor). Les anticorps utilisés sont décrits dans le tableau S2.
Analyse statistique
Toutes les données ont été représentées graphiquement et analysées à l'aide de GraphPad Prism 5 (GraphPad Software). Les données présentées sont la moyenne ± s.e.m. Des tests t de Student ont été utilisés pour la comparaison entre deux groupes. Les différences entre les groupes ont été analysées par une analyse de variance à sens unique (ANOVA) suivie du test de comparaisons multiples de Dunnett.
RÉSULTATS
Six lignées de cellules iPS humaines ont été utilisées dans cette étude, trois dérivées de contrôles sains (CT1, CT2 et CT3) et trois dérivées d'individus affectés par le WS avec un syndrome de Wolfram identifié génétiquement et cliniquement (WS1, WS2 et WS5). Les informations sur les iPSCs CT et WS sont résumées dans le tableau S1. Toutes les lignées cellulaires ont été validées à l'aide de tests classiques pour les cellules souches du cerveau (NSC) et les neurones post-mitotiques ont montré une absence de protéine wolframine contrairement à toutes les cellules CT (Figure 1 et Figure S1E).
Les neurones WS présentent des troubles de la croissance des neurites
Lorsque les lignées cellulaires CT et WS ont été différenciées, aucune différence n'a été observée entre les deux types de lignées en termes de détection des marqueurs de CSN, NESTIN et SOX2, et des marqueurs de neurones corticaux, HuC/D et CUX2, ainsi que du nombre total de cellules (Figure 1A, B, C, et Figure S1D). En revanche, l'immunomarquage de TUJ1 dans les neurites a révélé une différence morphologique frappante entre les cellules CT et WS. En effet, les neurones CT formaient un réseau stellaire typique de fibres fines et ramifiées alors que les cellules WS présentaient des processus longs et larges (jusqu'à plusieurs millimètres de longueur) sans ramification apparente (Figure 2A et Figure S2A, D). En utilisant la microscopie confocale, ces grands processus semblaient être formés par l'agrégation de neurites parallèles regroupés ensemble (Figure 2B). L'imagerie sur cellules vivantes a révélé que ces figures neuritiques anormales sont apparues entre les 10e et 14e jours de différenciation neuronale in vitro (DIV), selon la lignée cellulaire (films supplémentaires A, B, C et D). Dans les cultures neuronales WS, les faisceaux neuritiques se sont constitués progressivement alors que les neurites se développaient directement dans la culture sans interaction apparente avec les amas cellulaires. Les processus se sont allongés et agrandis pendant environ une semaine, après quoi les réseaux neuritiques sont restés stables. En outre, les extrémités en croissance de certains faisceaux neuritiques ont formé des demi-tours inhabituels et même des cercles complets (figure S2B).
Afin de quantifier le phénomène, nous avons mesuré l'épaisseur des neurites isolés et en faisceau dans les cultures CT et WS. Bien que la plupart des structures susmentionnées aient une taille inférieure à 3 µm dans les cellules CT et WS, les cellules WS présentaient un nombre plus faible de ces processus fins que les cellules CT. Inversement, le nombre de faisceaux de neurites était à la fois plus abondant et plus grand dans les cellules WS, atteignant jusqu'à 58 µm de largeur alors que les structures neuritiques CT ne dépassaient pas 12 µm. Par conséquent, l'épaisseur moyenne des structures neuritiques était plus élevée dans les neurones WS que dans les CT (Figure 2C). Pour comparer davantage les cellules WS et CT, nous avons défini trois intervalles d'épaisseur (0 à 3 µm, 3 à 6 µm et > 6 µm) et observé que >90% des structures neuritiques CT se situaient dans l'intervalle de 0 à 3 µm alors que cette proportion était significativement réduite à environ 80 % dans les cellules WS (Figure 2D et Figure S2C). Inversement, il y avait moins de 1 % de structures neuritiques par mm2 avec une épaisseur > 6 µm dans les cellules CT alors que ce nombre atteignait 10 % dans les cellules WS (Figure 2D, E et Figure S2C). Ces résultats indiquent que l'absence de wolframine entraîne une croissance neuritique aberrante qui pourrait avoir un impact sur la fonctionnalité neuronale.
La restauration de la wolframine normalise la croissance des neurites dans les neurones WS
Pour confirmer que l'absence de wolframine était impliquée dans le phénotype neuritique, une lignée de cellules iPS WS sauvées de WFS1 a été générée en utilisant l'édition de gènes CRISPR/Cas9 pour introduire l'ADNc de WFS1 sous un promoteur inductible par la doxycycline au locus AAVS1 (la lignée WS5 a été utilisée ; Figure 3A). Les caractéristiques pluripotentes et le caryotype normal ont été maintenus dans la lignée de cellules iPS sauvées (WS5R) (Figure S3A, B). Le succès de l'édition de gènes a été confirmé après un traitement à la doxycycline (Dox) à 50 ng/mL, car la synthèse de la wolframine a été détectée dans les CSN et les neurones (figure 3B). Les cellules WS5R se sont différenciées de manière similaire à la lignée WS5 parentale en NSCs (Figure S3C). De plus, l'expression de WFS1 lors du traitement avec 50 ng/mL de Dox n'a pas modifié le pourcentage de neurones HuC/D+ et CUX2+ (Figure 3C, D), indiquant ainsi que les propriétés initiales de la lignée cellulaire n'ont pas été modifiées par l'expression du transgène. En revanche, l'induction de l'expression de WFS1 par Dox a modifié le réseau neuritique (Figure 3E), le ramenant à ce qui avait été observé dans les cellules CT. Des réseaux stellaires typiques de neurites minces s'étendant entre les grappes de cellules ont été observés dans les cultures traitées par Dox. La quantification des neurites isolés et groupés par mm2 a montré une normalisation du profil qui ressemblait étroitement à celui des cultures CT (Figure 3G, H et Figure S3D). Nous avons observé des résultats similaires pour l'épaisseur moyenne (Figure 3F). À titre de contrôle, le traitement à la doxycycline de neurones WS5 non modifiés n'a pas affecté les marqueurs de différenciation et n'a pas non plus eu d'effet sur la croissance neuritique (Figure S3E, F, G), ce qui montre que la normalisation du réseau neuritique par le traitement à la doxycycline nécessite la présence de la construction WFS1.
Les cellules WS présentent des défauts dans l'expression des gènes impliqués dans le neurodéveloppement et le guidage des axones.
L'expression globale des gènes a été comparée entre les NSC et les neurones CT et WS. Dans une analyse en composantes principales (ACP), les échantillons se sont regroupés en fonction de leur phénotype (NSCs CT, NSCs WS, neurones CT et neurones WS) (figure 4A). En revanche, les deux groupes CT et WS différaient par le nombre de gènes différentiellement exprimés (DEG) (valeur P ≤ 1 % ; Fold Change≥1,5 ; minReads100). Il y avait 493 DEGs pour WS par rapport aux NSCs CT (249 upregulés et 244 downregulés) et 254 DEGs pour les neurones WS, par rapport aux neurones CT (87 upregulés et 167 downregulés). Parmi ces DEG, 29 étaient couramment dérégulés dans les NSC et les neurones WS (Figure S4A). Lorsque les DEG des neurones ont été mis en correspondance avec les voies de l'Encyclopédie des gènes et des génomes de Kyoto (KEGG), la voie la plus enrichie était celle du "guidage des axones" (figure 4B). L'annotation fonctionnelle a révélé que les trois catégories ontologiques de gènes les plus enrichies étaient "neurogenèse", "développement du système nerveux" et "guidage des axones" (figure 4B). Dans les cellules WS, les gènes liés à la croissance et au guidage des axones étaient soit régulés à la hausse, comme le roundabout-1 (ROBO1, OMIM 602430), soit régulés à la baisse, comme le Slit Guidance Ligand 3 (SLIT3, OMIM 603745), la semaphorine 4A (SEMA4A, OMIM 607292), la tenascine C (TNC, OMIM 187380) et plusieurs éphrines (EPH). Les DEG associés à ces catégories étaient également régulés à la baisse à l'état de CSN, comme le récepteur D de la nétrine Unc-5 (UNC5D, OMIM 616466), la protéine 43 associée à la croissance (GAP43, OMIM 162060), le récepteur 1 de l'éphrine de type B (EPHB1, OMIM 600600) ou la TNC (Figure S4B). Les résultats ont été confirmés pour un ensemble de gènes par qRT-PCR (Figure 4C). Dans l'ensemble, ces données suggèrent qu'un programme de croissance des neurites est dérégulé dans les neurones WS.
En tenant compte de la littérature qui associe les processus de dégénérescence dans le syndrome de Wolfram à l'activation de la réponse aux protéines mal conformées (UPR)11, nous avons spécifiquement analysé l'expression de certains gènes cibles de l'UPR. En condition basale (et véhicule), aucune différence entre les NSC CT et WS n'a été détectée (Figure S4C, D). En revanche, l'activation de la réponse au stress du RE en traitant les cellules avec 50 nM de thapsigargine (TG) a révélé un retour plus lent à la ligne de base de l'épissage de l'ARNm de la protéine 1 de liaison à la boîte X (XBP1, OMIM 194355) dans les cellules WS que dans les cellules CT, ce qui indique que les cellules WS pourraient être plus sensibles au stress ER (Figure 4D, E). Ces résultats suggèrent que la signalisation de l'UPR pourrait être affectée dans les cellules de WS et pourrait conduire à des altérations chroniques et de faible intensité de la signalisation, entraînant à leur tour des phénotypes pathologiques.
Sauvetage pharmacologique des neurones de WS et mécanismes d'action potentiels
Pour tester cette dernière hypothèse, nous avons cherché à étudier comment les trois branches de l'UPR pouvaient contribuer au phénotype neuritique identifié en utilisant des inhibiteurs pharmacologiques sélectifs. Grâce à cette approche, ni l'inhibition de l'IRE1α (à l'aide du MKC-8866) ni la modification de la réponse intégrée au stress (ISR, à l'aide du salubrinal) n'ont permis de modifier le réseau neuritique anormal des cellules WS (Figure 5A-F et Figure S5A-G). En revanche, l'inhibition de la branche ATF6α de l'UPR à l'aide de 10 µM de céapine-A7 a sauvé la formation de neurites par les cellules WS pendant la différenciation neuronale. Les cultures de cellules WS traitées à la céapine-A7 ont montré des réseaux neuritiques qui ressemblaient étroitement aux cultures CT en termes de nombre de structures neuritiques dans l'intervalle supérieur à 6 µm d'épaisseur (Figure 5G-I et Figure S5H-J) et d'épaisseur moyenne (2,162 +/- 0,256 µm pour NT contre 1,265 +/- 0,177 µm pour la ceapine-A7). Ces résultats suggèrent l'existence de mécanismes de signalisation ATF6 non conventionnels (non aigus) dans les cellules WS, susceptibles de conduire à des structures neuritiques aberrantes et qui sont atténués par la ceapine-A7.
Le traitement par l'acide valproïque a des effets positifs sur la croissance des neurites et les gènes de guidage de l'axone dans les neurones du WS
Dans le même ordre d'idées, les effets de différents médicaments actuellement testés chez des personnes atteintes du SS ou en cours d'étude préclinique, à savoir le dantrolène, modulateur du calcium, le liraglutide, agoniste du GLP1, l'acide 4-phénylbutyrique (4PBA), inhibiteur des HDAC, et l'acide valproïque (VPA, également décrit comme un atténuateur de la signalisation du stress du RE), ont été évalués sur le phénotype d'excroissance des neurites pathologiques. La neuraminidase (NM) a été utilisée comme témoin car cette enzyme réduit l'adhésion non spécifique entre les neurites. En conséquence, 10 mU de NM ont empêché la formation de fascicules dans les neurones WS lorsqu'ils ont été appliqués au début de la différenciation neuronale post-mitotique (Figure 6A). La NM n'a eu aucun effet sur l'expression globale des gènes. Il n'y avait aucune différence dans les défauts de croissance des neurites entre les cultures de cellules WS traitées et non traitées lorsqu'on utilisait soit 100 nM de liraglutide, 1 mM d'acide 4-phénylbutyrique ou 10 µM de dantrolène (figure S6A). En revanche, les cellules WS traitées par 1,5 mM de VPA ont montré une modification du phénotype neuritique anormal, affichant un profil comparable à celui des neurones WS traités par NM (figure 6A). Les neurones WS traités à cette dose ont présenté un réseau neuritique similaire à celui des neurones CT, tant en termes de fibres isolées que de fibres groupées (figure 6B, C, D et figure S6B). Parallèlement, le VPA a augmenté l'expression de SLIT3, TNC et EPHB1, trois des gènes de guidage axonal qui sont régulés à la baisse dans les cellules WS (figure 6E). Le VPA n'a eu aucun impact sur le nombre de cellules ou la proportion de neurones HuC/D+ dans les cultures, ce qui exclut un effet toxique (figure S6C, D). En revanche, les tentatives de correction du phénotype pathologique après son installation en commençant le traitement par VPA à DIV 18 ont été inefficaces. À la recherche d'indices supplémentaires concernant les mécanismes moléculaires, le VPA a été testé pour un effet protecteur sur la réponse prolongée de stress ER induite par la thapsigargin observée dans les NSC WS. Aucune protection n'a été obtenue avec ce médicament. L'acide valproïque a donc démontré un effet préventif sur l'anomalie de la croissance des neurites observée dans les neurones WS, dont le mécanisme n'est pas lié à une protection contre une réponse anormale au stress du RE.
DISCUSSION
Le principal résultat de cette étude est la démonstration que les neurones humains dérivés d'iPSC avec des mutations associées au syndrome de Wolfram présentent des défauts de développement neurologique phénotypiques et moléculaires. Cela se traduit par une altération de l'excroissance neuritique et du cheminement axonal, ainsi que par une fasciculation anormalement étendue en culture. Les changements dans l'expression génétique concernent un ensemble de gènes impliqués dans le neurodéveloppement et le cheminement axonal. On peut supposer que ces altérations pathologiques participent à la réduction du volume cérébral chez les individus atteints de WS. Bien que l'expression des gènes codant pour des protéines impliquées dans la réponse aiguë à l'UPR n'ait pas été modifiée, les cellules WS sont apparues plus sensibles au stress ER. Parmi les candidats thérapeutiques actuellement explorés, seul l'acide valproïque a démontré une capacité à atténuer certains des défauts de développement neurologique observés dans les neurones de WS.
Les résultats de notre étude apportent un éclairage sur une série de résultats cliniques qui semblaient jusqu'à présent paradoxaux. En effet, les altérations neurodégénératives progressives propres au syndrome de Wolfram qui affectent plusieurs voies neurosensorielles2 ; 7 ; 25, le tronc cérébral et le cervelet5 ; 26 n'apparaissent que plusieurs années après la naissance, dans l'enfance et l'adolescence, et progressent dans le temps. Cependant, ces phénomènes, qui ont des corrélations cliniques dramatiques, sont précédés d'une diminution globale du volume cérébral, tant dans la matière grise que dans la matière blanche, comme l'a récemment démontré une série d'études IRM3 ; 4. Ces altérations ont été observées très tôt dans la vie et n'ont pas évolué avec l'âge, suggérant un défaut de développement neurologique jusqu'ici ignoré car cliniquement silencieux12. Conformément à cette observation, des altérations neurodéveloppementales du cerveau ont également été observées dans des modèles animaux de la maladie27-29. Nos résultats de culture cellulaire in vitro pourraient bien fournir une base mécanistique pour ces altérations neurodéveloppementales précoces. En effet, une partie des neurones du syndrome de stress post-traumatique présentait des anomalies de croissance des neurites qui pourraient les rendre inaptes à l'élaboration d'ensembles normaux de connexions, ce qui pourrait avoir des conséquences sur la survie des neurones30. Les neurites épais en faisceau présentaient des trajectoires qui n'étaient pas similaires à celles observées dans les cultures de cellules CT, s'étendant sur de très longues distances sans attraction apparente vers les corps cellulaires et formant des courbes ou même des boucles complètes. L'imagerie time-lapse a révélé une altération concomitante des repères de guidage à l'extrémité des neurites en faisceau. Ces changements phénotypiques ont été associés de manière cohérente à une dysrégulation prévalente d'un ensemble de gènes appartenant au sous-ensemble "guidage des axones". Parmi les gènes les plus dérégulés figuraient SLIT3, SEMA4A, UNCD5, TNC, GAP43, ROBO1 et plusieurs EPHRINS, qui codent tous pour des protéines nécessaires au cheminement axonal et dont le défaut est associé à des anomalies neurodéveloppementales majeures31 ;32. Différentes études ont décrit un lien potentiel entre les défauts d'orientation axonale et la réduction du volume cérébral, à la fois dans des modèles murins et chez des personnes33-36. Les mécanismes précis par lesquels la dérégulation de l'expression de ces gènes peut conduire à une réduction du volume cérébral chez les personnes atteintes du syndrome de Wolfram restent cependant à établir complètement.
La plupart des gènes dérégulés mentionnés ci-dessus codent pour des protéines situées au niveau de la membrane plasmique ou exerçant leurs fonctions de manière extracellulaire. En tant que tels, ils nécessitent des voies de biogenèse fonctionnelles, qui englobent la voie sécrétoire. De manière intéressante, il a été démontré que la wolframine agit comme un régulateur principal de l'homéostasie du RE, à savoir la réponse aux protéines mal conformées, et les mécanismes pathologiques associés au syndrome de Wolfram ont été largement attribués à un contrôle défectueux de ces processus en raison de la perte de la wolframine10. Dans notre modèle, il n'y avait pas de différence entre l'expression au départ de l'un des marqueurs de stress ER aigu dans les cellules neurales dérivées des lignées de cellules iPS CT ou WS, contrairement à ce qui a été rapporté pour les cellules pancréatiques17 ; 37. Ce n'est que lorsque le stress du RE a été provoqué par la thapsigargine que les NSC WS ont montré une activation prolongée de l'épissage de l'ARNm XBP1 par rapport à la CT, ce qui indique une capacité réduite de ces cellules à faire face à un stress prolongé du RE. Ces résultats suggèrent que, même si dans des conditions basales, les marqueurs canoniques de stress du RE ne sont pas détectés, l'homéostasie du RE semble être affectée dans les cellules neurales WS. Cette hypothèse a été confirmée lorsque le phénotype associé à la maladie a été atténué par un traitement à la ceapine-A7, un inhibiteur de l'ATF638. Puisqu'il a été démontré que la wolframine inhibe l'activation de l'ATF610, nos résultats suggèrent que le développement des neurites peut être affecté par une activation constitutive/chronique de l'ATF6 dans les cellules WS, ce qui peut conduire à une altération de l'homéostasie du RE et de la capacité à gérer le stress dans ces cellules.
L'observation d'un phénotype pathologique clair associé à des marqueurs d'expression génétique altérés dans les cellules neuronales in vitro nous a incités à analyser les effets thérapeutiques potentiels des médicaments qui font actuellement l'objet d'études précliniques ou cliniques. Le paysage des traitements testés pour le syndrome de Wolfram est, en effet, particulièrement riche, favorisé par l'observation d'un certain nombre de défauts fonctionnels potentiellement traitables1 ; 39. La perte démontrée de l'homéostasie calcique des cellules 40 ; 41 a ainsi conduit à un essai clinique basé sur le dantrolène, un stabilisateur du calcium du RE (NCT02829268). Parallèlement, l'inhibiteur non sélectif de l'histone désacétylase, l'acide valproïque (VPA), fait actuellement l'objet d'un essai clinique (NCT03717909) après avoir démontré qu'il inversait la production défectueuse de P21cip dans les cellules knock-out WFS1 42. D'autres médicaments font l'objet d'études précliniques, notamment les agonistes du GLP1, comme le liraglutide, qui s'est révélé neuroprotecteur dans les modèles cellulaires de Wolfram, en plus de son effet attendu sur la sécrétion d'insuline43 ; 44, et la chaperone chimique à activité anti-oxydante 4-PBA39. Dans notre modèle, seul le VPA a démontré une action préventive dose-dépendante sur la croissance aberrante des neurites, et ceci était associé à la normalisation de l'expression des gènes codant pour les protéines de guidage des axones. Il a été démontré que le VPA réduit le stress du RE45, ce qui pourrait contribuer à ses effets positifs sur les neurones du WS. On peut également émettre l'hypothèse d'un effet moins spécifique puisque le VPA est un inhibiteur d'histone désacétylase non spécifique très puissant qui a un impact sur plus de mille gènes dans les cellules neuronales in vitro46. En tant que médicament neuroactif pléiotrope, il est connu pour exercer des effets neuroprotecteurs dans un large éventail de modèles de maladies neurodégénératives, notamment la sclérose latérale amyotrophique47, la maladie de Parkinson48 ou les lésions de la moelle épinière49. Les résultats de deux études qui ont exploré la promotion de la croissance des neurites par le VPA dans des modèles de défauts neuronaux dus à différentes mutations CMT, Rab7 dans le CMT2B50 et AARS dans le CMT2N51, sont particulièrement intéressants pour l'interprétation de nos données. Le fait que le VPA semble favoriser la croissance des neurites de manière similaire dans des modèles pathologiques basés sur des mutations génétiques différentes suggère fortement que ces effets médicamenteux ne sont pas spécifiques d'un mécanisme pathologique spécifique. Néanmoins, nos données soutiennent l'hypothèse selon laquelle le VPA pourrait contribuer à atténuer certains symptômes chez les personnes atteintes du syndrome de Wolfram, ce qui est actuellement testé en clinique.
La question de savoir si les défauts neurodéveloppementaux identifiés dans le cheminement axonal peuvent être associés après des années aux phénomènes neurodégénératifs observés ou si les processus neurodéveloppementaux et neurodégénératifs sont dus à des mécanismes pathologiques indépendants n'est à ce stade qu'une question ouverte. Il convient toutefois de mentionner qu'au cours des dix dernières années, plusieurs auteurs ont émis l'hypothèse d'un lien entre ces anomalies différentielles et d'autres maladies neurodégénératives à apparition tardive. Dans une étude fondamentale, Lin et ses collègues52 ont souligné le fait que les analyses de l'ensemble du génome de la maladie de Parkinson ont révélé que les polymorphismes d'un seul nucléotide dans les gènes de guidage de l'axone étaient prédictifs d’évolutions différentes de la maladie de Parkinson. Cette constatation a été étendue à d'autres maladies neurodégénératives53 et, en particulier, une altération du bourgeonnement axonal a été hypothétiquement associée à une dérégulation des protéines de guidage axonal dans la sclérose latérale amyotrophique. Le guidage axonal est fondamental pour l'établissement d'une connectivité et d'une fonctionnalité appropriées des neurones dans le système nerveux central adulte. Il a été proposé que la dérégulation des processus de développement nécessitant le guidage des axones puisse être compensée jusqu'à un certain point dans le temps mais conduise plus tard à la dégénérescence52-55. Inversement, des défauts subtils dans les protéines qui jouent un rôle non seulement pendant le neurodéveloppement mais aussi dans la neuroplasticité adulte peuvent contribuer à une neurodégénérescence retardée.
Données supplémentaires
Les données supplémentaires comprennent 6 figures, 4 films et 3 tableaux.
Remerciements
I-Stem fait partie de l'Institut des Biothérapies pour les Maladies Rares (IdB) soutenu par l'Association Française contre les Myopathies (AFM-Téléthon). Ce projet a également bénéficié de subventions de l'Association syndrome de Wolfram, de l'Association Nationale de la Recherche et de la Technologie et de l'Agence Nationale pour la Recherche : NeurATRIS ANR-11-INBS-0011 et Labex REVIVE ANR-10-LABX-73. M.-G.B.-M. a été soutenu par le programme "investissement d'avenir" INGESTEM. Nous remercions Yolande Masson et Lina El Kassar pour le caryotypage des lignées cellulaires et Alexandre Carteron pour le support technique des expériences d'ampliseq. Nous remercions le Dr Cécile Delettre et le regretté Pr Christian Hamel (Institut des neurosciences de Montpellier) pour le don des fibroblastes WS5, et la New York Stem Cell Foundation pour les lignées iPSC WS1 et WS2. Nous remercions le Pr Ole Isacson (Harvard Medical School) pour ses conseils et sa lecture du manuscrit et le Pr Nathalie Holic pour ses conseils en matière d'édition du génome. Nous reconnaissons avec gratitude le soutien du PSMN (Pôle Scientifique de Modélisation Numérique) de l'ENS de Lyon pour les ressources informatiques.
Déclaration d'intérêts : Les auteurs ne déclarent pas d'intérêts concurrents.
Disponibilité des données et des codes : Les ensembles de données transcriptomiques générés au cours de cette étude sont disponibles au NCBI GEO sous le numéro d'accession GSE156911 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156911).
RESSOURCES WEB
REFERENCES
$11. Pallotta, M.T., Tascini,G.,Crispoldi, R., Orabona,C.,Mondanelli, G., Grohmann,U.,and Esposito,S.(2019).Wolframsyndrome,arareneurodegenerativedisease:frompathogenesistofuturetreatmentperspectives. Journal of translationalmedicine 17,238.
$12. Hershey,T.,Lugar,H.M.,Shimony,J.S.,Rutlin,J.,Koller,J.M.,Perantie,D.C.,Paciorkowski,A.R.,Eisenstein,S.A.,Permutt,M.A.,andWashingtonUniversityWolframStudy,G.(2012).Earlybrain vulnerabilityin Wolframsyndrome.PloSone7,e40604.
$13. Lugar,H.M.,Koller,J.M.,Rutlin,J.,Marshall,B.A.,Kanekura,K.,Urano,F.,Bischoff,A.N.,Shimony,J.S.,Hershey,T.,andWashingtonUniversityWolframSyndromeResearchStudy,G.(2016).NeuroimagingevidenceofdeficientaxonmyelinationinWolframsyndrome.SciRep6,21167.
$14. Lugar,H.M.,Koller,J.M.,Rutlin,J.,Eisenstein,S.A.,Neyman,O.,Narayanan,A.,Chen,L.,Shimony,J.S.,andHershey,T.(2019).Evidenceforalteredneurodevelopmentandneurodegenerationin Wolframsyndrome using longitudinal morphometry. Sci Rep 9,6010.
$15. Shannon,P.,Becker,L.,andDeck,J.(1999).EvidenceofwidespreadaxonalpathologyinWolframsyndrome. Acta neuropathologica98,304-308.
$16. Chaussenot,A.,Bannwarth,S.,Rouzier,C.,Vialettes,B.,Mkadem,S.A.,Chabrol,B.,Cano,A.,Labauge,P.,andPaquis-Flucklinger,V.(2011).Neurologicfeaturesandgenotype-phenotypecorrelation inWolframsyndrome. Annalsof neurology 69,501-508.
$17. Barrett,T.G.,Bundey,S.E.,andMacleod,A.F.(1995).Neurodegenerationanddiabetes:UKnationwidestudyofWolfram(DIDMOAD) syndrome.Lancet346,1458-1463.
$18. Rohayem,J., Ehlers,C.,Wiedemann, B.,Holl,R.,Oexle,K.,Kordonouri, O.,Salzano, G.,Meissner,T.,Burger, W.,Schober, E.,etal. (2011).Diabetes and neurodegeneration inWolframsyndrome:a multicenterstudyof phenotype and genotype. Diabetescare34,1503-1510.
$19. Takeda,K.,Inoue,H.,Tanizawa,Y.,Matsuzaki,Y.,Oba,J.,Watanabe,Y.,Shinoda,K.,andOka,Y.(2001).WFS1(Wolframsyndrome1)geneproduct:predominantsubcellularlocalizationtoendoplasmicreticuluminculturedcellsandneuronalexpressioninratbrain.Humanmoleculargenetics10, 477-484.
$110. Fonseca,S.G.,Ishigaki,S.,Oslowski,C.M.,Lu,S.,Lipson,K.L.,Ghosh,R.,Hayashi,E.,Ishihara,H.,Oka,Y.,Permutt,M.A., etal. (2010). Wolframsyndrome1 gene negatively regulates ER stresssignaling in rodent and human cells. TheJournalof clinical investigation120,744-755.
$111. Fonseca,S.G.,Fukuma,M.,Lipson,K.L.,Nguyen,L.X.,Allen,J.R.,Oka,Y.,andUrano,F.(2005).WFS1isanovelcomponentoftheunfoldedproteinresponseandmaintainshomeostasisoftheendoplasmicreticuluminpancreaticbeta-cells.TheJournalofbiologicalchemistry280,39609-39615.
$112. Samara,A.,Rahn,R.,Neyman,O.,Park,K.Y.,Samara,A.,Marshall,B.,Dougherty,J.,andHershey,
T. (2019). Developmental hypomyelination in Wolfram syndrome: new insights fromneuroimaging andgene expression analyses.Orphanet journalof rare diseases14, 279.
$113. Chambers,S.M.,Fasano,C.A.,Papapetrou,E.P.,Tomishima,M.,Sadelain,M.,andStuder,L.(2009).HighlyefficientneuralconversionofhumanESandiPScellsbydualinhibitionofSMADsignaling. Nature biotechnology 27,275-280.
$114. Boissart,C.,Nissan,X.,Giraud-Triboult,K.,Peschanski,M.,andBenchoua,A.(2012).miR-125potentiatesearlyneuralspecificationofhumanembryonicstemcells.Development139,1247-1257.
$115. Boissart,C.,Poulet,A.,Georges,P.,Darville,H.,Julita,E.,Delorme,R.,Bourgeron,T.,Peschanski,M.,andBenchoua,A.(2013).Differentiationfromhumanpluripotentstemcellsofcorticalneuronsofthesuperficiallayersamenabletopsychiatricdiseasemodelingandhigh-throughput drug screening.Translational psychiatry3,e294.
$116. Darville,H.,Poulet,A.,Rodet-Amsellem,F.,Chatrousse,L.,Pernelle,J.,Boissart,C.,Heron,D.,Nava,C.,Perrier,A.,Jarrige,M.,etal.(2016).HumanPluripotent Stem Cell-derivedCorticalNeuronsforHighThroughputMedicationScreeninginAutism:AProofofConceptStudyinSHANK3HaploinsufficiencySyndrome.EBioMedicine 9,293-305.
$117. Shang,L.,Hua,H.,Foo,K.,Martinez,H.,Watanabe,K.,Zimmer,M.,Kahler,D.J.,Freeby,M.,Chung,W.,LeDuc,C.,etal.(2014).beta-celldysfunctionduetoincreasedERstressinastemcellmodelof Wolframsyndrome.Diabetes63, 923-933.
$118. Yu,J.,Hu,K.,Smuga-Otto,K.,Tian,S.,Stewart,R.,Slukvin,II,andThomson,J.A.(2009).Humaninduced pluripotentstem cells freeofvector andtransgene sequences. Science324,797-801.
$119. Hetz,C.,Axten,J.M., and Patterson,J.B.(2019).Pharmacologicaltargetingoftheunfoldedproteinresponsefordisease intervention. Nature chemicalbiology 15,764-775.
$120. Wakade, C.G.,Mehta,S.H., Maeda,M.,Webb, R.C., andChiu, F.C.(2013).Axonal fasciculation andtheroleofpolysialicacid-neuralcelladhesionmoleculeinratcorticalneurons.Journalofneuroscienceresearch 91,1408-1418.
$121. Li,W.,Turner,A.,Aggarwal,P.,Matter,A.,Storvick,E.,Arnett,D.K.,andBroeckel,U.(2015).ComprehensiveevaluationofAmpliSeqtranscriptome,anoveltargeted whole transcriptomeRNA sequencingmethodology for global geneexpression analysis.BMCgenomics 16, 1069.
$122. Amrhein,V.,Greenland,S.,andMcShane,B.(2019).Scientistsriseupagainststatisticalsignificance. Nature567,305-307.
$123. Chen,E.Y.,Tan,C.M.,Kou,Y.,Duan,Q.,Wang,Z.,Meirelles,G.V.,Clark,N.R.,andMa'ayan,A.(2013).Enrichr:interactiveandcollaborativeHTML5genelistenrichmentanalysistool.BMCbioinformatics14,128.
$124. Kuleshov,M.V.,Jones,M.R.,Rouillard,A.D.,Fernandez,N.F.,Duan,Q.,Wang,Z.,Koplev,S.,Jenkins,S.L.,Jagodnik,K.M.,Lachmann,A.,etal.(2016).Enrichr:acomprehensivegenesetenrichmentanalysis web server 2016 update. Nucleic acids research 44, W90-97.
$125. Carson,M.J.,Slager,U.T.,and Steinberg, R.M. (1977). Simultaneousoccurrenceof diabetesmellitus,diabetesinsipidus,andopticatrophyinabrotherandsister.Americanjournalofdiseasesofchildren 131,1382-1385.
$126. Scolding,N.J.,Kellar-Wood,H.F.,Shaw,C.,Shneerson,J.M.,andAntoun,N.(1996).Wolframsyndrome:hereditarydiabetesmellituswithbrainstemandopticatrophy.Annalsofneurology39,352-360.
$127. Cagalinec,M.,Liiv,M.,Hodurova,Z.,Hickey,M.A.,Vaarmann,A.,Mandel,M.,Zeb,A.,Choubey,V.,Kuum,M.,Safiulina,D.,etal.(2016).RoleofMitochondrialDynamicsinNeuronalDevelopment:Mechanismfor WolframSyndrome.PLoS biology14, e1002511.
$128. Plaas,M.,Seppa,K.,Reimets,R.,Jagomae,T.,Toots,M.,Koppel,T.,Vallisoo, T.,Nigul,M.,Heinla,I.,Meier,R.,etal.(2017).Wfs1-deficientratsdevelopprimarysymptomsofWolframsyndrome:insulin-dependentdiabetes,opticnerveatrophyandmedullarydegeneration.SciRep 7,10220.
$129. Sakakibara,Y.,Sekiya,M.,Fujisaki,N.,Quan,X.,andIijima,K.M.(2018).Knockdownofwfs1,aflyhomologofWolframsyndrome1,inthenervoussystemincreasessusceptibilitytoage-andstress-inducedneuronaldysfunctionanddegenerationinDrosophila.PLoSgenetics14,e1007196.
$130. Zweifel,L.S.,Kuruvilla, R., and Ginty,D.D.(2005). Functionsand mechanismsofretrogradeneurotrophin signalling. Nature reviewsNeuroscience6, 615-625.
$131. Wang,L.,andMarquardt,T.(2013).Whataxonstelleachother:axon-axonsignalinginnerveandcircuit assembly. Currentopinion in neurobiology 23,974-982.
$132. Roig-Puiggros,S.,Vigouroux,R.J.,Beckman,D.,Bocai,N.I.,Chiou,B.,Davimes,J.,Gomez,G.,Grassi,S.,Hoque,A.,Karikari,T.K.,etal.(2020).Constructionandreconstructionofbraincircuits:normalandpathological axon guidance. J Neurochem 153,10-32.
$133. Engle,E.C.(2010).Humangeneticdisordersofaxonguidance.ColdSpringHarborperspectivesinbiology2,a001784.
$134. Hibar,D.P.,Stein,J.L.,Jahanshad,N.,Kohannim,O.,Hua,X.,Toga,A.W.,McMahon,K.L.,deZubicaray,G.I.,Martin,N.G.,Wright,M.J.,etal.(2015).Genome-wideinteractionanalysisrevealsreplicatedepistaticeffectsonbrainstructure.Neurobiologyofaging36Suppl 1,S151-158.
$135. Vosberg,D.E.,Zhang,Y.,Menegaux,A.,Chalupa,A.,Manitt,C.,Zehntner,S.,Eng,C.,DeDuck,K.,Allard,D.,Durand,F.,etal.(2018).MesocorticolimbicConnectivityandVolumetricAlterationsinDCCMutationCarriers.TheJournalofneuroscience:theofficialjournaloftheSocietyforNeuroscience38,4655-4665.
$136. Deloulme,J.C.,Gory-Faure,S.,Mauconduit,F.,Chauvet,S.,Jonckheere,J.,Boulan,B.,Mire,E.,Xue,J.,Jany,M.,Maucler,C.,etal.(2015).Microtubule-associatedprotein6mediatesneuronalconnectivity throughSemaphorin3E-dependentsignallingforaxonalgrowth.Naturecommunications6,7246.
$137. Urano,F.(2014).WolframsyndromeiPScells:thefirsthumancellmodelofendoplasmicreticulumdisease.Diabetes63,844-846.
$138. Gallagher,C.M.,andWalter,P.(2016).CeapinsinhibitATF6alphasignalingbyselectivelypreventingtransportof ATF6alphato the Golgi apparatus duringER stress. eLife5.
$139. Abreu,D.,andUrano,F.(2019).Current LandscapeofTreatmentsforWolframSyndrome.Trendsin pharmacologicalsciences 40, 711-714.
$140. Lu,S.,Kanekura,K.,Hara,T.,Mahadevan,J.,Spears,L.D.,Oslowski,C.M.,Martinez,R.,Yamazaki-Inoue, M., Toyoda, M.,Neilson,A., etal. (2014).A calcium-dependent proteaseasapotentialtherapeutictargetforWolframsyndrome.ProceedingsoftheNationalAcademyofSciencesof theUnitedStatesof America111, E5292-5301.
$141. Cagalinec,M.,Zahradnikova,A.,Zahradnikova,A.,Jr.,Kovacova,D.,Paulis,L.,Kurekova,S.,Hot'ka,M.,Pavelkova,J., Plaas,M.,Novotova, M.,etal.(2019).CalciumSignalingandContractilityinCardiac MyocyteofWolframine Deficient Rats. Frontiers in physiology10,172.
$142. Gharanei,S.,Zatyka,M.,Astuti,D.,Fenton,J.,Sik, A.,Nagy,Z.,andBarrett,T.G.(2013).Vacuolar-typeH+-ATPaseV1AsubunitisamolecularpartnerofWolframsyndrome1(WFS1)protein,whichregulates itsexpression andstability. Human molecular genetics 22,203-217.
$143. Kondo, M.,Tanabe, K., Amo-Shiinoki, K.,Hatanaka,M., Morii, T.,Takahashi,H.,Seino, S.,Yamada,Y.,andTanizawa,Y.(2018).ActivationofGLP-1receptorsignallingalleviatescellularstressesandimprovesbetacellfunctioninamousemodelofWolframsyndrome.Diabetologia61,2189-2201.
$144. Seppa,K.,Toots,M.,Reimets,R.,Jagomae,T.,Koppel,T.,Pallase,M.,Hasselholt,S.,KrogsbaekMikkelsen,M.,RandelNyengaard,J.,Vasar,E.,etal.(2019).GLP-1receptoragonistliraglutidehas a neuroprotectiveeffectonan aged ratmodel ofWolframsyndrome.Sci Rep9,15742.
$145. Li,Z.,Wu,F.,Zhang,X.,Chai,Y.,Chen,D.,Yang,Y.,Xu,K.,Yin,J.,Li,R.,Shi,H.,etal.(2017).ValproateAttenuatesEndoplasmicReticulumStress-InducedApoptosisinSH-SY5YCellsviatheAKT/GSK3beta Signaling Pathway. International journal of molecularsciences18.
$146. Fukuchi,M.,Nii,T.,Ishimaru,N.,Minamino,A.,Hara,D.,Takasaki,I.,Tabuchi,A.,andTsuda,M.(2009).Valproicacidinducesup-ordown-regulationofgeneexpressionresponsiblefortheneuronal excitation andinhibitionin ratcortical neuronsthrough itsepigenetic actions.Neuroscienceresearch 65,35-43.
$147. Wang,X.,Ma,M.,Teng,J.,Che,X.,Zhang,W.,Feng,S.,Zhou,S.,Zhang,Y.,Wu,E.,andDing,X.(2015).ValproateAttenuates25-kDaC-TerminalFragmentofTDP-43-InducedNeuronalToxicityviaSuppressingEndoplasmicReticulumStressandActivatingAutophagy.Internationaljournal ofbiological sciences 11,752-761.
$148. Harrison,I.F.,Crum, W.R.,Vernon,A.C.,and Dexter,D.T.(2015).NeurorestorationinducedbytheHDACinhibitorsodiumvalproateinthelactacystinmodelofParkinson'sisassociatedwithhistoneacetylationandup-regulationofneurotrophicfactors.Britishjournalofpharmacology172,4200-4215.
$149. Chu,T.,Zhou,H.,Lu,L.,Kong,X.,Wang,T.,Pan,B.,andFeng,S.(2015).Valproicacid-mediatedneuroprotectionandneurogenesisafterspinalcordinjury:frommechanismtoclinicalpotential.Regenerativemedicine 10,193-209.
$150. Yamauchi,J.,Torii,T.,Kusakawa,S.,Sanbe,A.,Nakamura,K., Takashima,S.,Hamasaki,H.,Kawaguchi,S.,Miyamoto,Y.,andTanoue,A.(2010).ThemoodstabilizervalproicacidimprovesdefectiveneuriteformationcausedbyCharcot-Marie-Toothdisease-associatedmutantRab7through the JNKsignaling pathway. Journalof neuroscienceresearch88,3189-3197.
$151. Tatsumi,Y.,Matsumoto,N.,Iibe,N.,Watanabe,N.,Torii,T.,Sango,K.,Homma,K.,Miyamoto,Y.,Sakagami,H.,andYamauchi,J.(2019).CMTtype2N disease-associatedAARSmutantinhibitsneurite growththat can bereversed byvalproic acid.Neuroscienceresearch 139, 69-78.
$152. Lin,L.,Lesnick,T.G.,Maraganore,D.M.,andIsacson,O.(2009).Axonguidanceandsynapticmaintenance:preclinicalmarkersforneurodegenerativediseaseandtherapeutics.TrendsNeurosci32, 142-149.
$153. VanBattum,E.Y.,Brignani,S.,andPasterkamp,R.J.(2015).Axonguidanceproteinsinneurologicaldisorders.The LancetNeurology 14,532-546.
$154. Stoeckli,E. (2017).Where does axon guidanceleadus? F1000Research 6,78.
$155. Schwamborn,J.C.(2018).IsParkinson'sDiseaseaNeurodevelopmentalDisorderandWillBrainOrganoids HelpUs toUnderstandIt? Stemcells anddevelopment27, 968-975.
TITRES ET LÉGENDES DES FIGURES
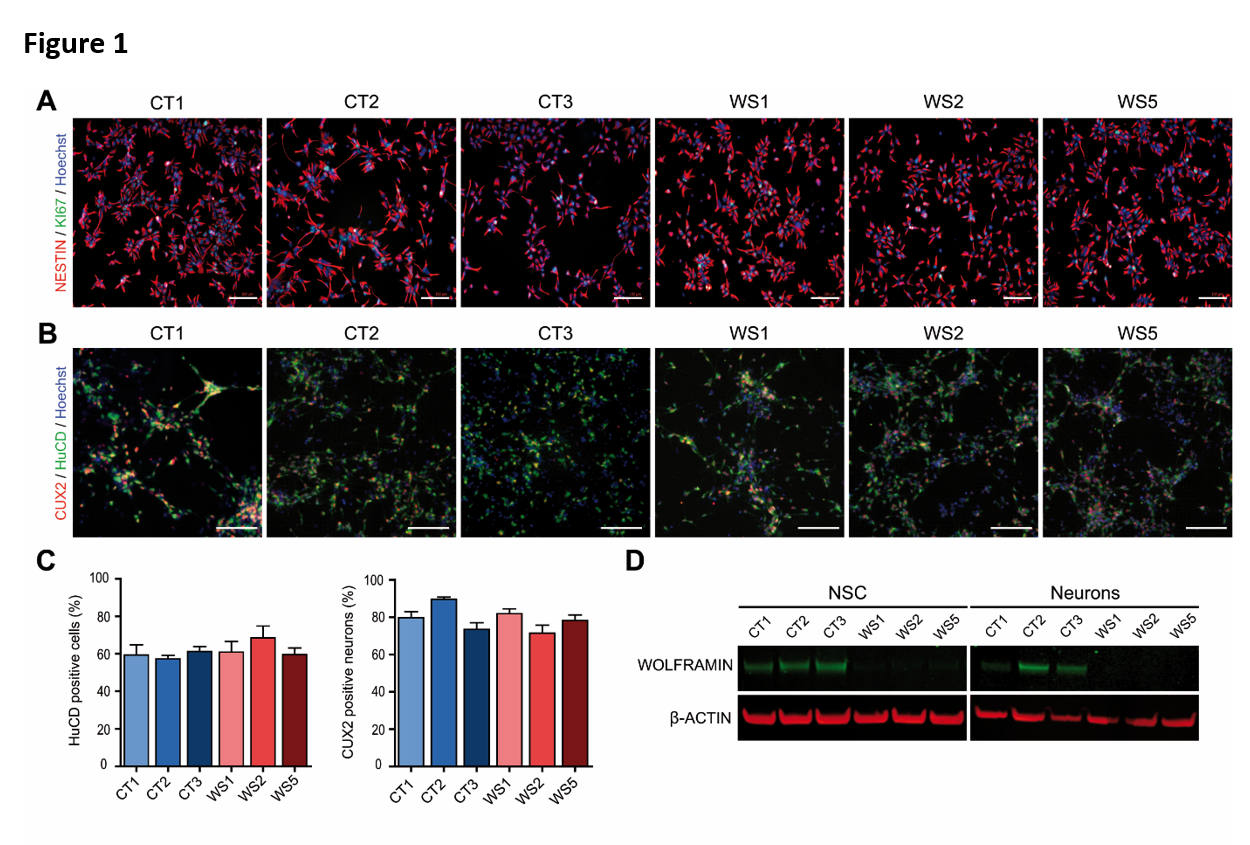
Figure 1 : Les iPSCs WS et CT génèrent toutes deux efficacement des populations homogènes de cellules souches neurales et de neurones. A. Microscopie par immunofluorescence pour NESTIN (marqueur de CSN) et Ki67 (marqueur de prolifération) dans les CSN dérivées des trois lignées iPSC de contrôle (CT1, CT2 et CT3) et des trois lignées iPSC WS (WS1, WS2, WS5). Barres d'échelle, 100 μm. B. Neurones corticaux immunoréactifs HuC/D et CUX2 au 14e jour de différenciation in vitro (DIV) à partir du stade NSC. Barres d'échelle, 100 μm. C. Proportion de neurones HuC/D+ et CUX2+-immunoréactifs dans la culture cellulaire à DIV 14 du stade NSC. Les données sont présentées sous forme de moyenne ± s.e.m. (n = 3 différenciations biologiquement indépendantes. Aucune différence statistique n'a été observée en utilisant l'ANOVA à sens unique avec le test post hoc de Dunnett). D. Western blot de la wolframine dans les NSC et les neurones à DIV 21.
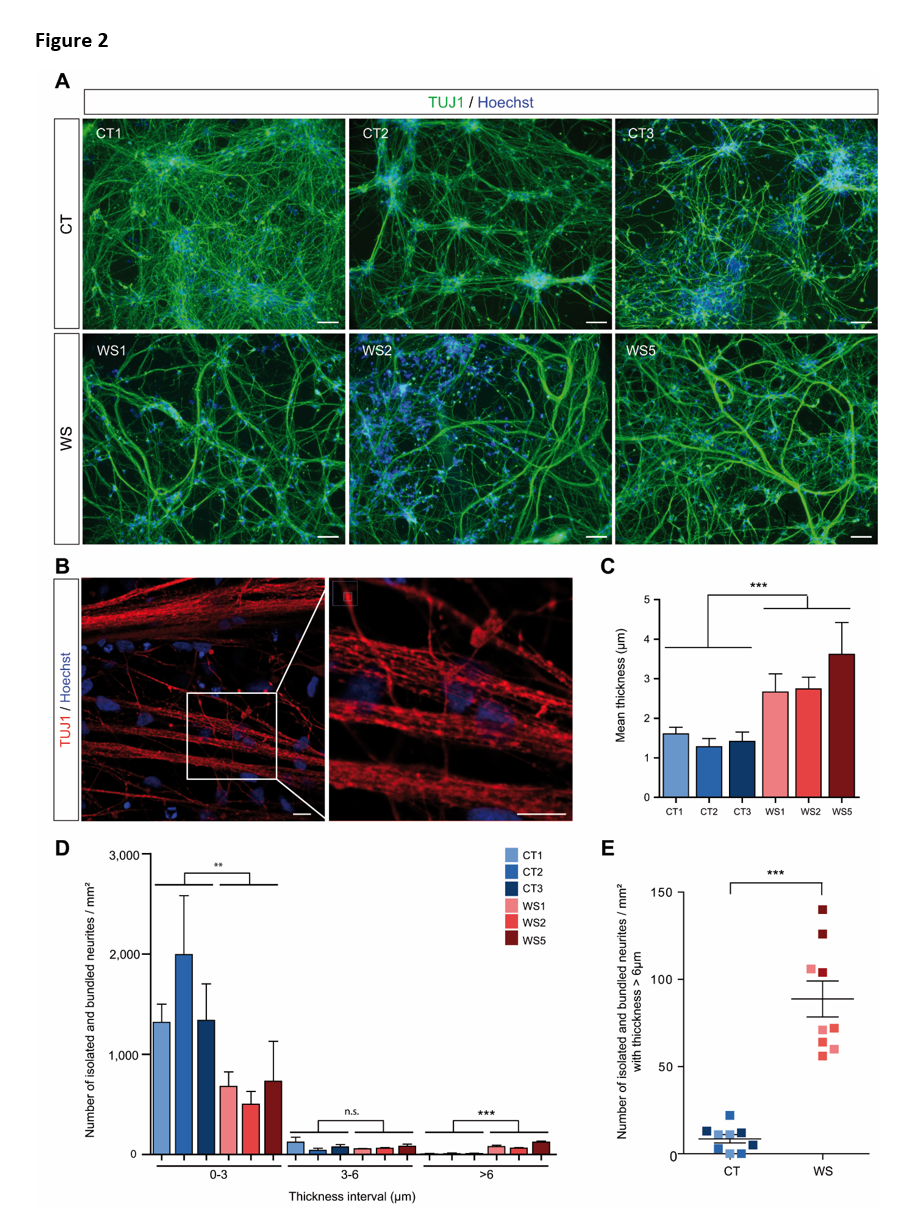
Figure 2 : Les neurones WS présentent une croissance anormale des neurites. A. Neurones CT et WS immunoréactifs au TUJ1 à la DIV 21. Barres d'échelle, 100 μm. B. Analyse confocale des neurones WS5 immunoréactifs au TUJ1 à la DIV 21. Barres d'échelle, 10 μm. C. Quantification de l'épaisseur moyenne des neurites isolés et en faisceau dans les neurones CT et WS. D. Quantification du nombre de neurites isolés et en faisceau par mm² sur trois intervalles d'épaisseur (0 - 3 ; 3 - 6 et > 6 µm) pour les neurones CT et WS (voir méthodes). E. Quantification du nombre moyen de neurites isolés et groupés pour les neurones CT et WS dans l'intervalle d'épaisseur > 6
µm d'épaisseur. Le code couleur indiquant les différentes lignées cellulaires est commun à c, d et e. Toutes les données sont présentées sous forme de moyenne ± s.e.m. (n = 3 différenciations biologiquement indépendantes ; **p < 0,01, ***p
< 0,001 ; test t de Student).
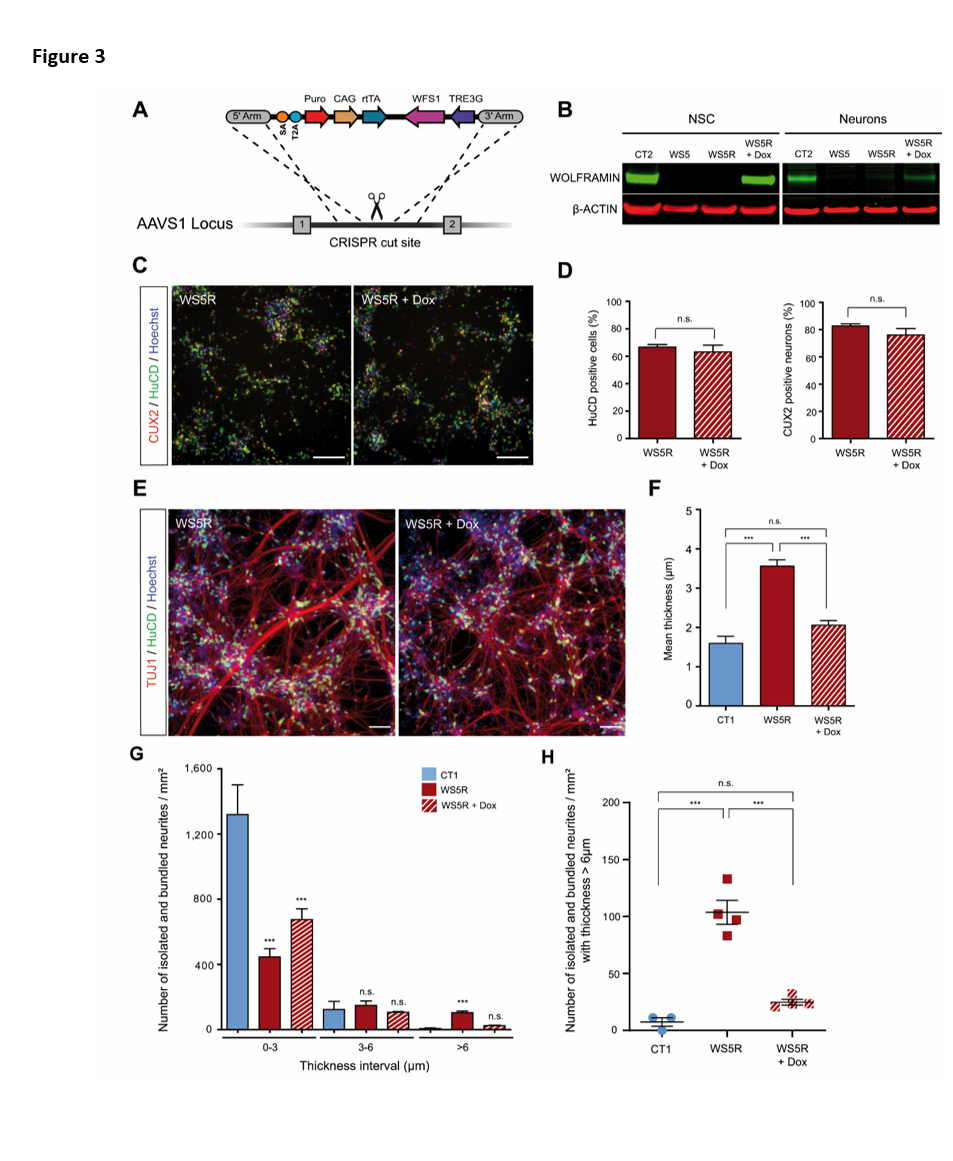
Figure 3 : Le knock-in de l'ADNc de WFS1 résout le phénotype des neurones de WS à DIV21. A. Stratégie d'édition de gènes utilisant la technologie CRISPR-Cas9 pour l'insertion au locus AAVS1 de l'ADNc de WFS1 sous un promoteur inductible par la doxycycline dans la lignée iPSC WS5. B. Détection par Western blot de la wolframine dans les NSC et les neurones WS5 (WS5R) sauvés avec ou sans traitement à la doxycycline (Dox) et dans CT2. C. Immunomarquage HuC/D et CUX2 des neurones WS5R traités ou non par Dox. Barres d'échelle, 100 μm. D. Proportion de cellules HuC/D+ et CUX2+-immunoréactives dans les neurones WS5R traités ou non au Dox. Les données sont présentées sous forme de moyenne ± s.e.m. (n = 3 différenciations biologiquement indépendantes ; test t de Student). E. Neurones WS5R immunoréactifs TUJ1 et HuC/D traités ou non par Dox. Barres d'échelle, 100 μm. F. Quantification de l'épaisseur moyenne des neurites isolés et en faisceau pour les neurones WS5R traités ou non au Dox par rapport au CT1. G. Quantification du nombre de neurites isolés et groupés par mm² pour trois intervalles d'épaisseur (0 - 3 ; 3 - 6 et > 6 µm) dans les neurones. H. Analyse du nombre de neurites isolés et groupés par mm² pour l'intervalle > 6 µm dans les neurones WS5R traités ou non par Dox par rapport à CT1. Pour f, g et h, les données sont exprimées par rapport à celles des cellules CT1 et présentées sous forme de moyenne ± s.e.m. (n=3 pour CT1 et n=4 pour les différenciations biologiquement indépendantes WS5R et WSR5 + Dox ; ***p < 0,001 ; ANOVA à sens unique avec test post hoc de Dunnett, les données sont comparées aux cellules CT1).
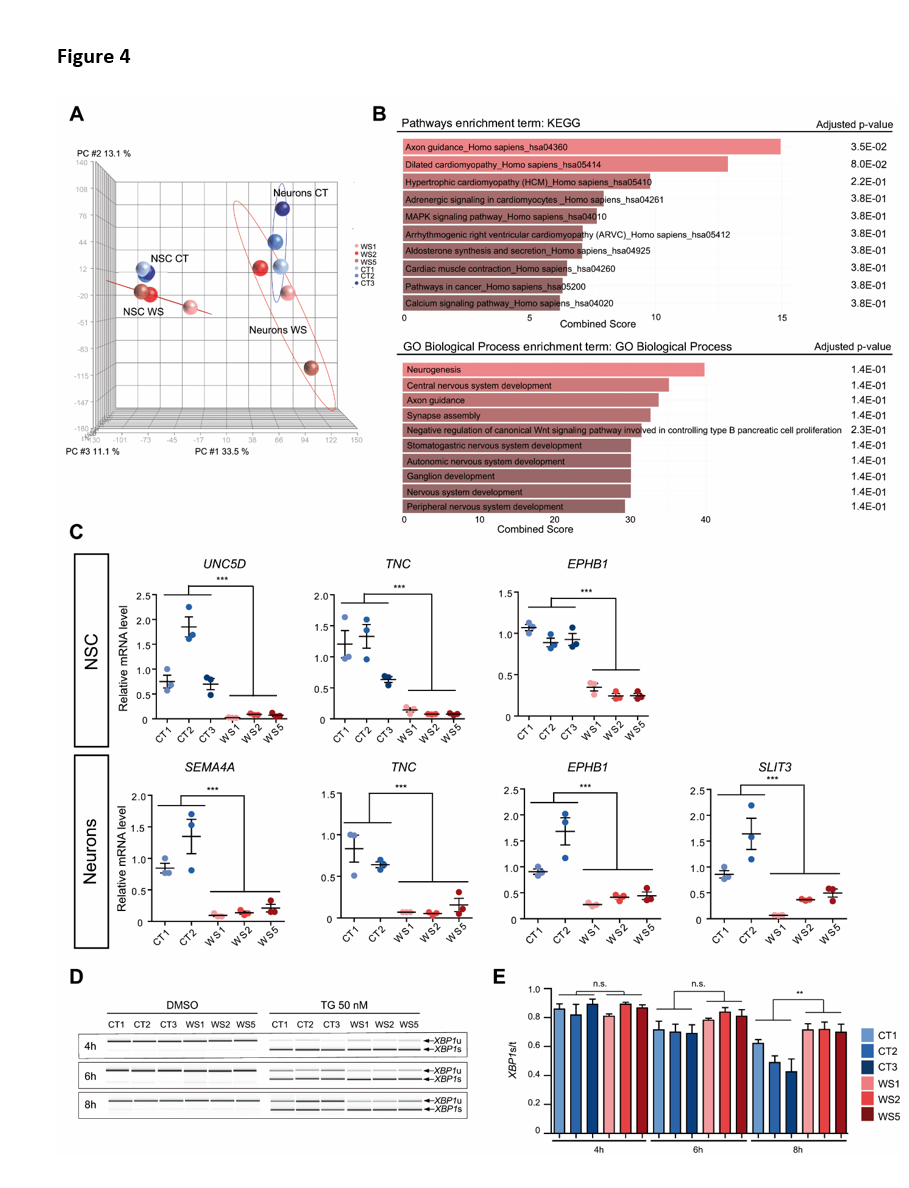
Figure 4 : Comparaison de l'expression génique globale entre les NSC et les neurones CT et WS. A et B.
Analyse du transcriptome à l'aide d'AmpliSeq. A. Analyse en composantes principales (ACP), entre les NSC et les neurones CT et WS.
NSCs et neurones. B. Analyse d'enrichissement des voies KEGG (panneau supérieur) et de l'ontologie des gènes (panneau inférieur) dans les neurones à l'aide de l'outil Enrichr. C. Analyse RT-PCR quantitative des niveaux d'ARNm des DEG dans les neurones et les NSC CT et WS. Les données sont exprimées par rapport à celles des cellules CT1 et normalisées par rapport à l'expression de l'ARNr 18S. Les données sont présentées sous forme de moyenne ± s.e.m. (n = 3 différenciations biologiquement indépendantes ;
***p < 0,001 ; test t de Student). D et E. Induction du stress ER dans les NSC CT et WS après 4, 6 et 8 heures de traitement à la thapsigargin (TG) 50 nM. D. Analyse de l'expression de XBP1 épissé (XBP1s) et non épissé (XBP1u). E. Quantification de l'expression de XBP1s normalisée par rapport au XBP1 total (XBP1t). Les données sont présentées sous forme de moyenne ± s.e.m. (n = 3 expériences biologiquement indépendantes ; *p<0,05 ; ANOVA à sens unique avec test post hoc de Dunnett).








