






---
Si vous voulez des renseignements sur les essais cliniques en cours :
vous pouvez nous écrire sur
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
---
Si vous voulez envoyer un courrier postal à l'association ou faire un don par chèque
Association du syndrome de Wolfram
Résidence Gauguin
56390 GRAND-CHAMP
---
vendredi 5 décembre :
journée des association de la filière Sensgene

---
mercredi 3 décembre :
Journée annuelle de la filières Firendo

un beau programme pour cette journée annuelle à l'hôpital Cochin à Paris.
---
vendredi 28 novembre :
Envoi d'un questionnaire orthoptie
Ce questionnaire a pour objectif de mieux comprendre l'impact de la maladie sur le quotidien,
d'évaluer les besoins spécifiques et les prises en charge actuels.
Il a été conçu par 3 étudiantes en orthoptie à l'université de Bordeaux.
---
samedi 15 novembre :
Réunion annuelle des familles

---
vendredi 14 novembre :
Assemblée générale de l'association

---
mardi 14 octobre :
8ème édition de la "coupe Wolfram"
au golf d'Isabelle, à Plaisir (78)

Merci à tous ceux qui contribuent à ce rendez-vous solidaire,
incontournable dans notre agenda.
---
mercredi 8 octobre :
Congrès RARE Paris 2025

Les rencontres RARE sont devenues un rendez-vous incontournable
pour tous les acteurs de la communauté des maladies rares.
Organisé tous les deux ans par la Fondation Maladies Rares,
cet événement offre un espace de réflexion centré plus particulièrement
sur la recherche dans toutes les dimensions du parcours du patient.
---
lundi 6 octobre :
Réunion du bureau
![]()
---
jeudi 2 octobre :
Date limite du dépôt des dossiers pour notre appel à projets

---
mercredi 1er octobre :
Journée mondiale contre le syndrome de Wolfram
![]()
---
mardi 30 septembre :

Nous intervenons dans une table ronde sur le rôle des associations de patients
dans la recherche contre les maladies rares, à l'invitation de la Fondation Maladies Rares.
---
samedi 20 septembre :
Festival Donner Du Sens à Sadirac (33)


---
mardi 29 juillet :
Lancement d'un nouvel appel à projets
Nous sommes heureux de vous annoncer que nous lançons un nouvel appel à projets pour soutenir la recherche à hauteur de 200 000 euros sur 2 ans.
Il est intégralement financé par notre association et nous tenons à remercier tous les donateurs et tous les bénévoles sans qui ce nouveau projet ne serait pas possible.
Un grand merci à la Fondation Maladies Rares pour son soutien précieux qui nous assure une large diffusion et une expertise de qualité des dossiers que nous recevrons.
---
jeudi 24 juillet :
Point d'étape avec le Dr. Pascal Reynier (Angers)
Le Dr. Pascal Reynier (en bas à gauche de la photo) nous expose l'étude métabolomique qu'il mène avec son équipe à Angers.
Il nous présente à la fois, les résultats à date mais aussi les perspectives à venir.
En présence du Dr. Orssaud (en haut à droite) et du Dr. Rossignol et d'Antoine Poulignon (en bas à droite) de l'équipe bordelaise.
---
lundi 30 juin :
Point d'étape avec l'équipe du Dr. Rodrigue Rossignol (Bordeaux)
De gauche à droite, Antoine Poulignon et le Dr. Rodrigue Rossignol.
En raison de l’impact multi-systémique de ce syndrome, des approches approfondies comme la protéomique sont essentielles pour éclaircir sa pathologie complexe et explorer des pistes thérapeutiques viables. L’analyse bioinformatique menée par Antoine Poulignon (Lauréat du Master 2 Bioinformatique de L’université de Lille ; en stage à U1211 à Bordeaux) sur les données de protéomique plasmatique obtenues à la plateforme ‘Bordeaux Protéome’ a révélé l’existence de trois marqueurs. Ce travail va se poursuivre par une thèse en septembre 2025, dirigée par le Dr. Rodrigue ROSSIGNOL (DR1 Inserm), qui étudiera le rôle biologique et physiopathologique de ces trois marqueurs en collaboration avec plusieurs experts nationaux dont les docteurs Christophe Orssaud et Pascal Reynier. Le sujet de thèse vise à approfondir l’analyse physiopathologique du syndrome de Wolfram, l’impact des mutations récessives et dominantes, et à découvrir de nouveaux mécanismes biochimiques et des pistes thérapeutiques potentielles.
---
dimanche 29 juin :
Concert à Saint Aunès (34)
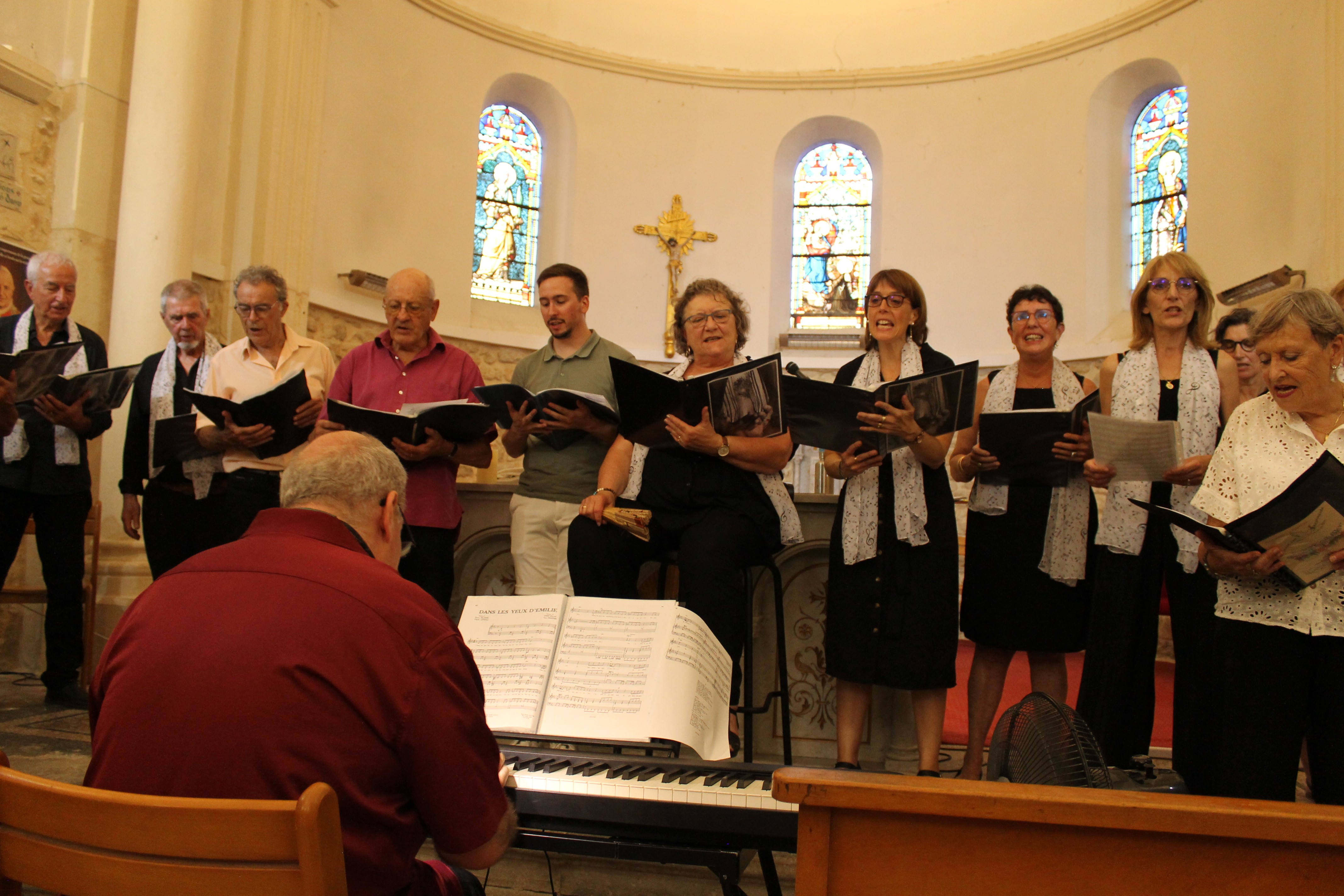
A St Aunes (34), la chorale de St Aunes et celle de Ribeyrolle de Jacou ont donné de la voix pour porter la nôtre.
Merci à toutes les chanteuses, à tous les chanteurs, aux bénévoles et à tout le public qui a pu profiter d'un beau concert ce dimanche 29 juin.
---
vendredi 27 juin :
Réunion avec les familles et 3 scientifiques
pour un résumé du congrès :

---
vendredi et samedi 13 et 14 juin :
10ème congrès mondial sur le syndrome de Wolfram
Nous avons organisé le dixième congrès mondial sur le syndrome de Wolfram.
Le vendredi au Sénat et le samedi à la faculté de pharmacie.
Près de 50 participants, 12 pays représentés et des présentations de haut vol qui donneront lieu à de belles collaborations pour faire avancer la recherche.
Merci au sénateur Yves Bleunven pour son accueil et à tous les chercheurs pour leur travail sans lequel aucun espoir n'est possible.
Depuis 2009 où nous étions partis d'une feuille blanche et où nous organisions le premier congrès mondial sur le syndrome de Wolfram,
la première rencontre des chercheurs, le nombre d'équipes a bien grandi
et la session complète sur les essais cliniques ce vendredi après-midi symbolise bien le chemin parcouru.
Forts de ces avancées et plus motivés que jamais, nous continuons le combat.
---
Retrouvez le magazine annuel
vous pouvez cliquer directement pour le consulter :
---
Magazine de l'INSERM de septembre
Article sur les travaux de Benjamin Delprat
cliquez sur l'image pour le lire
|
|
 |








